Introducing IFLAI CHROMA-1
We're introducing IFLAI CHROMA-1, the first model in our new CHROMA family of foundation models for phenotypic screening.
CHROMA-1 turns microscopy images into general-purpose embeddings that help scientists compare cellular states, connect genetic and chemical perturbations, and uncover biological relationships at scale. On the RxRx3-core biological relationship recall benchmark, CHROMA-1 achieves the top aggregate retrieval score while using substantially less training data, compute, and model scale than leading methods.
Compared with prior state-of-the-art models, CHROMA-1 uses 1,000-2,400x less compute, 3.2-18.6x less data, and 6-37x fewer trainable parameters, while setting a new aggregate benchmark score.
Why microscopy embeddings matter
Cell images contain rich information about how biological systems respond to perturbation. With high-throughput phenotypic screening, microscopy can be standardized and scaled to measure how cells respond to millions of chemical and genetic interventions, such as drug candidates, gene knockouts, or pathway modulators.
The challenge is turning these images into representations that are useful across experiments, instruments, cell types, and biological questions.
CHROMA-1 addresses this by encoding microscopy channels into meaningful vector representations. These embeddings make it possible to compare cellular phenotypes, map perturbations into a shared high-dimensional space, and apply downstream data science methods to study biological mechanism, target biology, and potential therapeutic activity.
What is CHROMA-1?
CHROMA stands for Cross-batch Harmonized Representations of Morphology with Active Sampling.
CHROMA-1 combines a modern ConvNeXt-based architecture with training strategies designed specifically for high-content biological imaging. The model is built to learn robust morphological representations while minimizing the technical variation that often limits phenotypic screening workflows.
A central part of CHROMA-1 is its active learning training strategy. In standard self-supervised learning, models can spend substantial compute on samples that add little new information. CHROMA-1 instead uses an active training loop that prioritizes the most informative training pairs.
This selection process is guided by a decision module that ranks candidate samples based on model uncertainty, biological relevance, representativeness, and the observed impact of similar samples in prior training rounds. By focusing training on examples most likely to improve the learned representation, CHROMA-1 increases sample efficiency and reduces unnecessary computation.
CHROMA-1 was trained on the compound subset of the CPG0016 Cell Painting dataset, comprising approximately 5 million images, using only 50 million trainable parameters.
Built for cross-batch robustness
Batch effects are one of the central challenges in high-content phenotypic screening. Differences in microscopes, staining conditions, plate layouts, operators, acquisition dates, and laboratory environments can introduce systematic shifts in image statistics that are unrelated to biology.
When these effects are not addressed, representations may cluster by acquisition batch rather than by phenotype. This can reduce reproducibility, obscure true biological signal, and limit transfer across instruments, sites, or assay conditions.
CHROMA-1 is designed to suppress batch-specific signals during representation learning while preserving biologically meaningful structure. Its architecture and training procedure incorporate mechanisms for cross-batch harmonization and domain adaptation, reducing the need for additional post hoc batch correction in downstream workflows.
The result is a model that maintains strong performance across experimental contexts and is better suited for deployment in real-world screening environments.
State-of-the-art retrieval on RxRx3
We evaluate CHROMA-1 in a zero-shot setting on the RxRx3-core biological relationship recall benchmark. This benchmark measures how well learned perturbation representations recover known biological relationships between genetic perturbations.
For each model, perturbation representations are compared using cosine similarity. These similarities are then evaluated against known relationships from public biological databases, including CORUM, hu.MAP, Reactome, and StringDB. The benchmark measures whether known relationships are enriched among the most similar perturbation pairs.
CHROMA-1 achieves the highest aggregate retrieval score across these databases, with a score of 0.419. This exceeds the previous SOTA models, Phenom-2 at 0.413 and Phenom-1 at 0.353.
What makes this result especially notable is the efficiency profile. CHROMA-1 reaches this level of performance without relying on massive training scale, using significantly less data and fewer trainable parameters. This efficiency translates into only 20 GPU-hours of training compute, compared with 20,000–48,000 GPU-hours for Phenom-1 and Phenom-2.
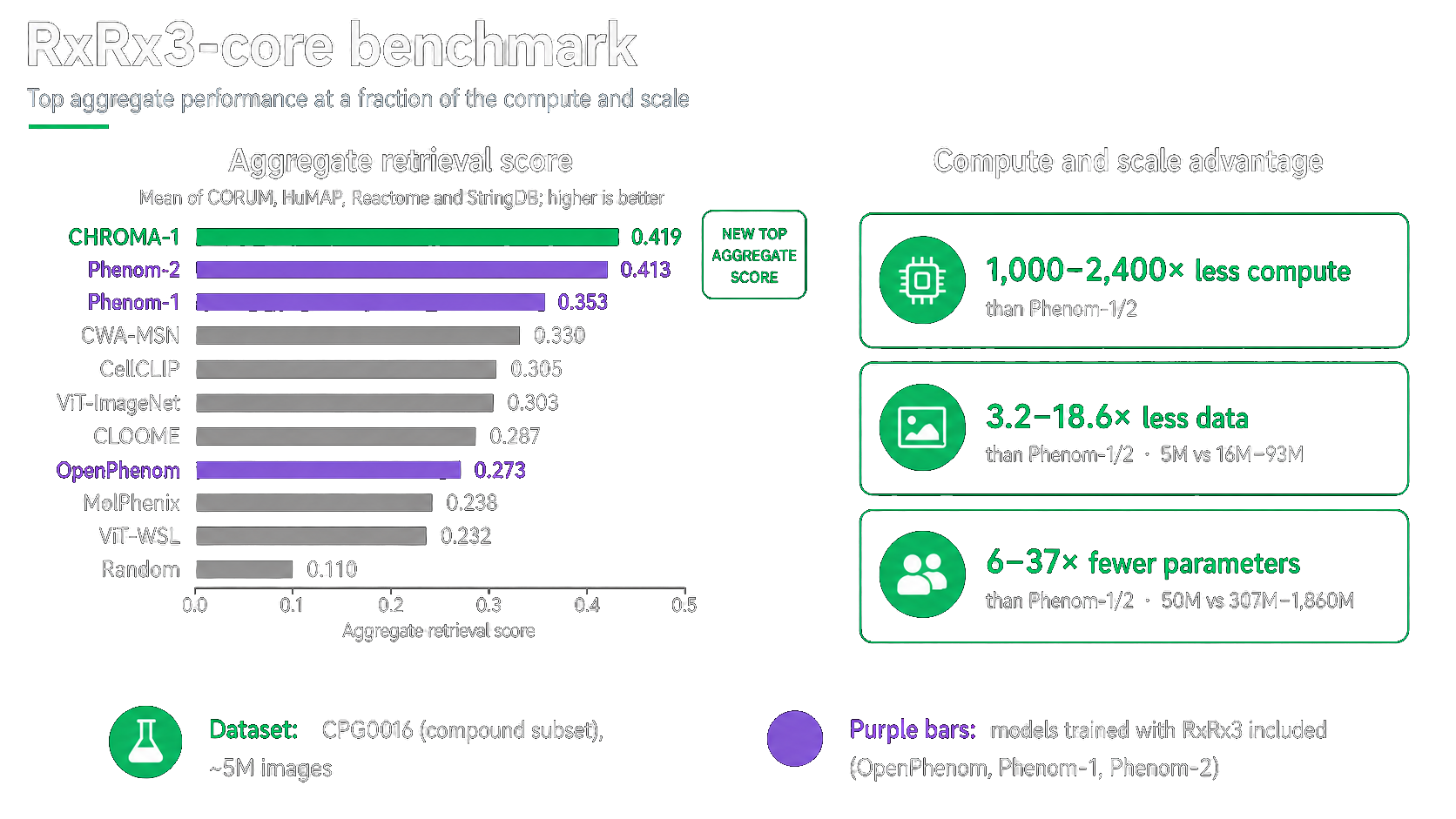
In other words, CHROMA-1 does not achieve its performance by simply scaling compute, data, or parameter count. It achieves top aggregate performance through architecture, active sampling, and cross-batch representation learning.
Designed for deployment
CHROMA-1 was built to support practical deployment across organizations.
In real screening programs, models must generalize across microscopes, laboratories, acquisition protocols, and cell types. CHROMA-1's domain adaptation and cross-batch robustness are designed to reduce performance degradation under these shifts.
For organizations with specialized assays or proprietary datasets, CHROMA-1 also supports efficient fine-tuning. Its training loops are designed to adapt to new domains with minimal additional data and compute, making it easier to customize the model for specific biological contexts.
Getting started
CHROMA-1 is the first release in the IFLAI CHROMA family. Our goal is to make high-performance phenotypic representations more accessible, efficient, and reliable for biological discovery.
Interested in CHROMA-1?
To learn more about deploying CHROMA-1 in your organization, contact us or register your interest and we will follow up with details on access, evaluation, and integration.